
随着现代临床试验越来越多地纳入远程元素、自适应研究设计和多来源数据,临床研究管理的复杂性持续增加。全面的数据和风险监测技术应运而生,研究团队为维持大型数据集质量,以及数据的真实性,也在采用越来越多的工具和方法,作为传统临床试验中基于研究中心的监查活动的补充。
传统的监查方法通常包括完整的研究中心审计和监查活动,往往借助于大量的人工源数据审阅(SDR)和源数据验证(SDV)进行。这两种方式不仅效率低下,也难以在全球范围的项目中应用。传统的方法由于停留在研究中心层面,缺乏研究水平整体视角,因此最后呈现的只能是独立的数据质量问题和中心层面的风险概况。一旦涉及到对研究整体水平数据风险进行早期检测,比如中心之间不良事件(AE)的差异性上报,仅仅依靠传统的单中心监查方法是无法完成的。
不良事件识别必须快狠准
在临床试验,尤其是国际多中心研究中,不良事件报告的差异性是一道反复出现的难题。研究表明,有多重原因导致不同国家的不良事件报告率存在差异,例如对研究方案和研究要求理解的差异,较轻微的事件判断是否上报的语言或文化差异,甚至包括不良事件上报引发的的社会污名观念等。
总体而言,不良事件报告的差异可能对患者安全和临床试验数据完整性产生负面影响。报告不充分可能耽误对患者安全重大新风险的识别,并影响药物安全档案的准确性和完整性。过度报告则可能因为混淆诊断与体征和症状,导致干扰数据的引入,增加非必要的人工数据审阅的需求,延迟对安全风险的检测。准确、完整、及时的不良事件报告对于评估和管理研究的安全数据来说至关重要,将对试验期间的关键决策产生重要影响。
不良事件报告的中心化统计监查
分析和统计方法可以用来客观识别跨安全领域的报告模式差异。这些分析能够识别研究中的数据离群值,并让用户(包括中心化监查员、数据管理人员或任何负责安全监查的人员)在研究中心或远程监查问题之前专注于深入调查部分试验中心。这种具备详细背景信息的分析视角能够帮助用户更早地接收到风险信号,并优化监查资源的分配。具有统计学显著性的不良事件报告差异信号可以被标记,以便监查团队进一步调查其根本原因,触发研究中心再培训,并确保研究中心具备准确和及时收集安全信息的能力。
评估时应考虑以下数据审查标准:
| 标准 | 考量因素 |
| 患者人数、事件数量和总体药物暴露 | 考虑到试验的高密度招募以及所有患者都处于试验早期阶段的情况(参见图1) |
| 合并用药的报告模式 | 临床试验研究中心的合并用药数量是否显著高于其他中心?是否存在没有相关不良事件和病史的合并用药? |
| 病史 | 病史信息是否充足?过多或过少?是否存在健康状况更为良好的患者,单指事件或缺失信息较少的患者? |
| 报告模式 | 未充分报告事件类型是否存在一定模式,例如轻微事件? |
| 患者人群 | 依据受影响的患者或患者亚群评估信息(参见图2) |
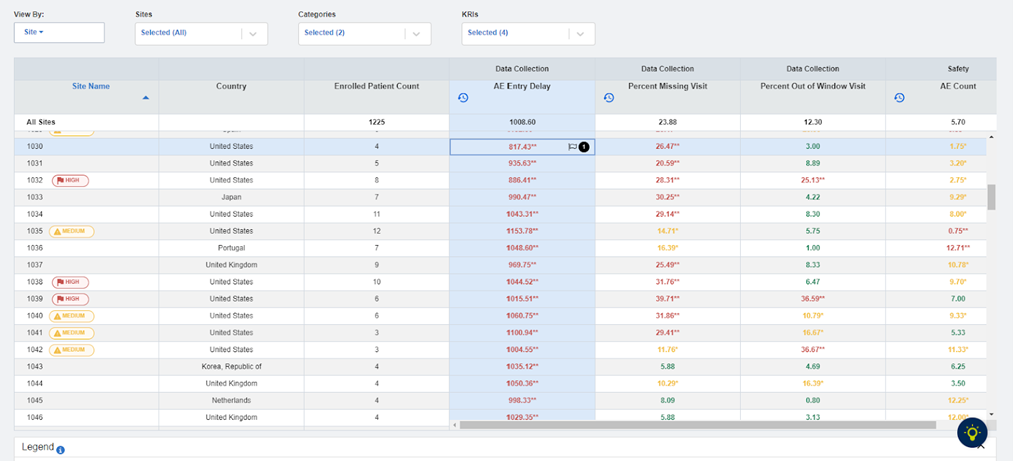
| 数据时效性 | 不良事件报告的差异是否由尚未录入的信息积压导致?注意特别评估不良事件录入延迟的关键风险指标(参见图3) |
图1:不良事件的未充分报告和过度报告
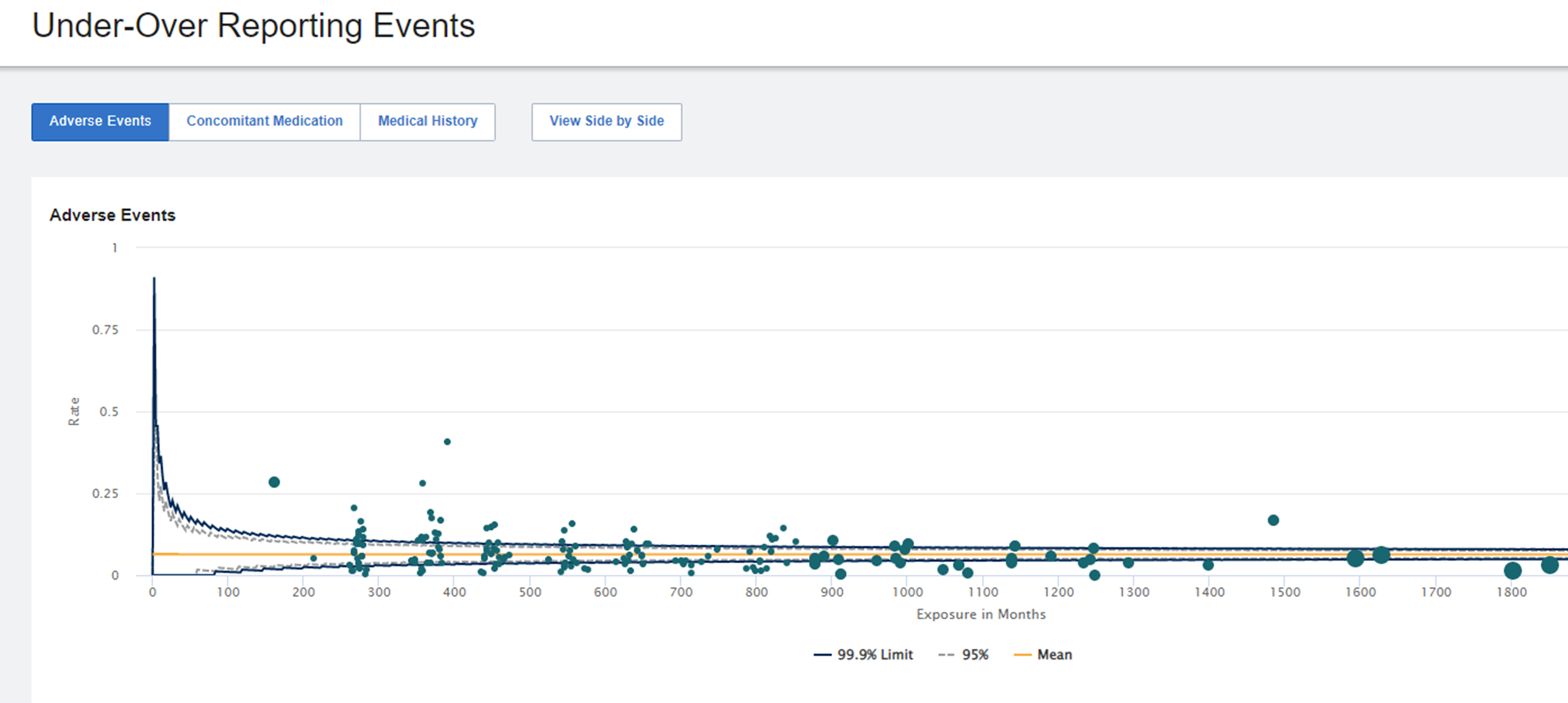
图2:患者档案——安全时间线表格
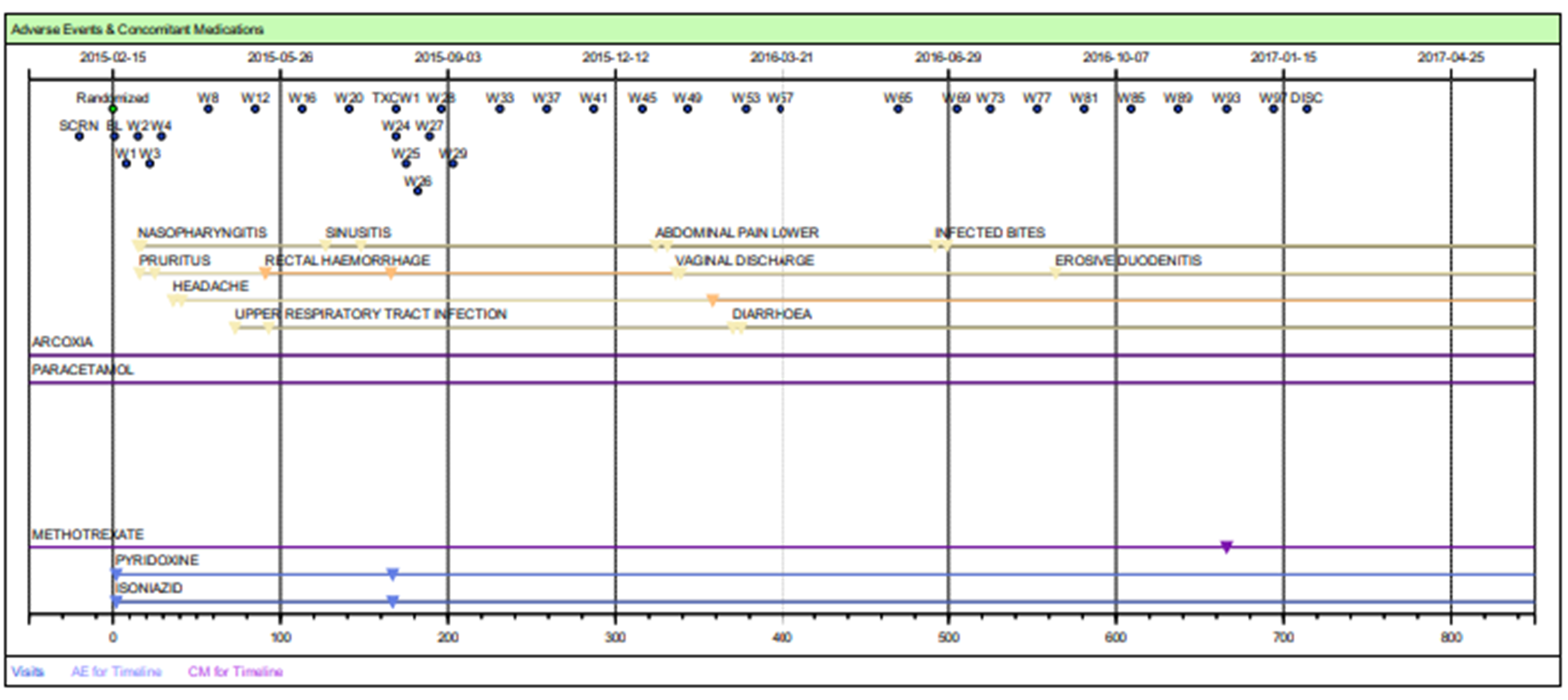
图3:关键风险指标仪表板:AE录入延迟度量指标
总结
传统的临床试验监查流程往往无法清晰体现报告模式的差异,诸如不良事件未充分报告这样的情况,往往需要进行跨研究中心评估以识别潜在风险信号。借助数据和监测技术,结合先进的分析方法、直观的可视化和来自所有患者数据源的实时数据,Medidata Detect能够确保充分的数据和风险监测,以及全面且及时的安全报告。
Medidata Detect是一款端到端的数据监测和中心化监查解决方案,允许数据管理和临床运营团队监测和限制影响患者安全和数据完整性的风险。这有助于从电子病历报告表(eCRF)和非电子病历报告表来源整合安全信息(不局限于不良事件数据集,还包括所有安全信息收集相关的外部数据),并作为不良事件报告评估的一部分进行交叉检查,包括实验室数据、患者报告结局、传感器数据和影像等。
详情请下载白皮书(英文版):


Contact Us
